Xenopus laevis egg extracts recapitulate complex genome maintenance processes that have not been reconstituted with purified components. Using this approach, we have discovered new DNA replication and repair mechanisms that are defective in cancer. Click on an area of study to learn more.
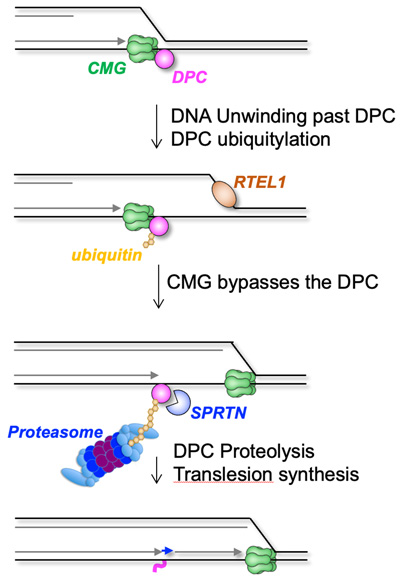
DNA-Protein Cross-Link Repair

Single molecule movie of CMGs bypassing DPCs
DNA-protein cross-links are generated by endogenous formaldehyde and other agents. Failure to repair DPCs causes aging and liver cancer. We discovered a new mechanism of DPC repair in which the replication fork triggers DPC proteolysis. Remarkably, the CMG helicase bypasses the DPC before proteolysis is activated, which we propose prevents inadvertent CMG proteolysis.
Selected Publications
Larsen et al. (2019). Replication-coupled DNA-protein crosslink repair by SPRTN and the proteasome in Xenopus egg extracts. Molecular Cell 73, 574-588.
Sparks et al. (2019). The CMG helicase bypasses DNA protein cross-links to facilitate their repair. Cell 176, 167-181.
Duxin et al. (2014). Replication-coupled repair of a DNA-protein crosslink. Cell 159, 346-357.
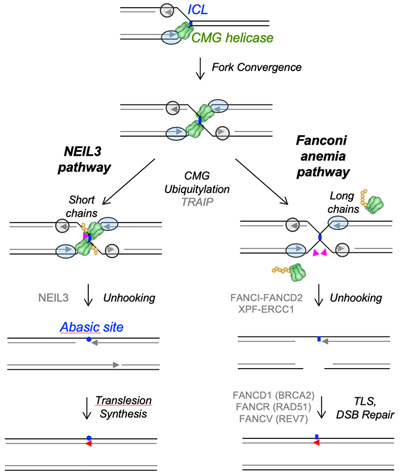
DNA Inter-Strand Cross-Link Repair
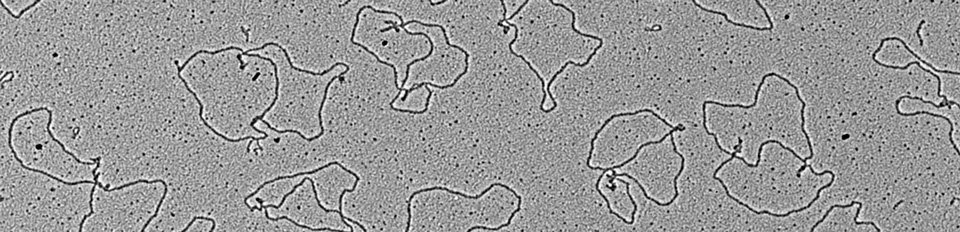
Electron micrograph of DNA undergoing ICL repair in egg extract
DNA inter-strand cross-links are caused by endogenous metabolites and cancer chemotherapeutics, and their repair is crucial to suppress cancer and bone marrow failure. Using replication of plasmids containing site-specific ICLs in egg extracts, we have characterized two distinct ICL repair pathways, both of which are triggered when replication forks converge on the lesion.
Selected Publications
Wu et al. (2019). TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 567, 267-272.
Semlow et al. (2016). Replication-dependent unhooking of DNA interstrand cross-links by the NEIL3 glycosylase. Cell 167, 498-511.
Knipscheer et al.. (2009). The Fanconi anemia pathway promotes replication-dependent DNA interstrand crosslink repair. Science 326, 1698-1701.
Räschle et al.(2008). Mechanism of replication-coupled DNA interstrand cross-link repair. Cell 134, 969-980.
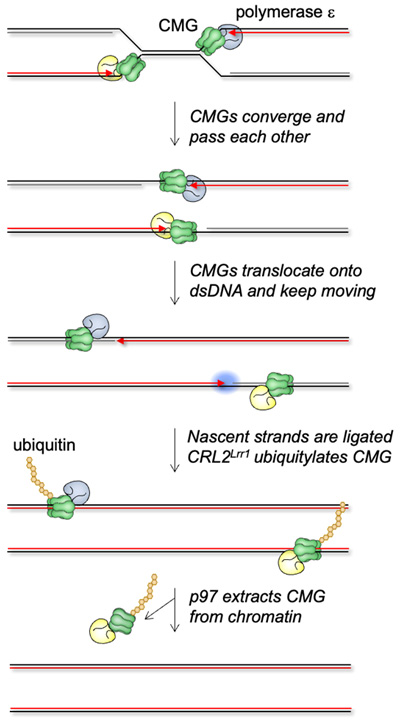
Replication Termination

Single molecule movie of CMGs converging and being unloaded from DNA
When replisomes from adjacent origins of replication converge, DNA synthesis must be completed and the replication machinery unloaded. By colliding replication forks with a lac repressor array and then releasing the array with IPTG, we have elucidated the molecular events underlying replication termination.
Selected Publications
Vrtis et al (2021). Single-strand DNA breaks cause replisome disassembly. Molecular Cell 81, 1309-1318.
Low et al. (2020). The replication fork suppresses CMG unloading from chromatin before termination. Genes&Development 34, 1534-45.
Deng et al. (2019). Mitotic CDK promotes replisome disassembly, fork breakage, and complex DNA rearrangements. Molecular Cell 73, 915-929.
Dewar et al. (2015). The mechanism of DNA replication termination in vertebrates. Nature 525, 345-350.
Transcription-Coupled Repair
Preparation of nucleoplasmic egg extract
RNA polymerases frequently collide with replisomes and DNA damage, but how these conflicts are resolved remains mysterious. We have succeeded in activating transcription in egg extracts (T. Mevissen, unpublished results), and we are interested in recapitulating transcription-coupled DNA repair in this system.